An integrative biology approach to model human skull, meningeal, and vascular malformations
Tischfield et al., Developmental Cell 2017.
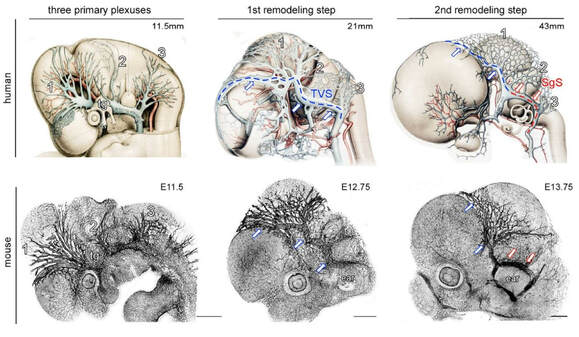
The growth and remodeling of the human (top) and mouse (bottom) dural cerebral vasculature is remarkably similar
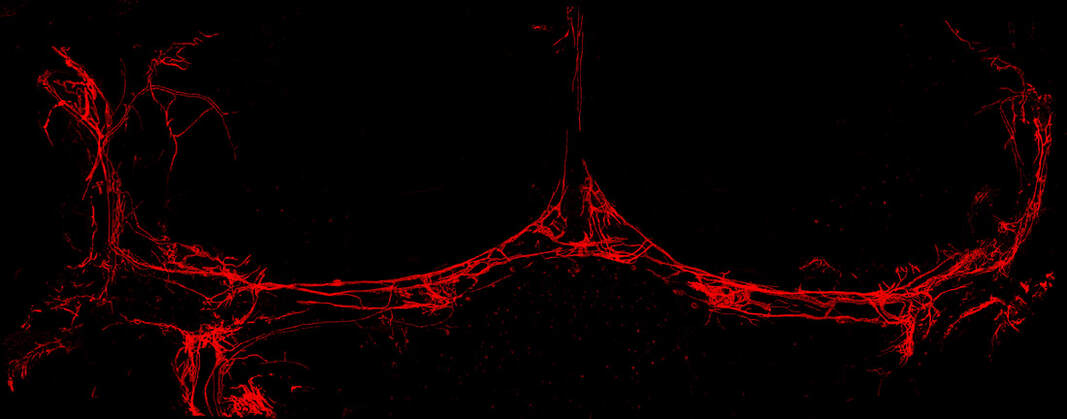
A prox1-tdTomato transgenic mouse depicting meningeal lymphatic networks along the venous sinuses
|
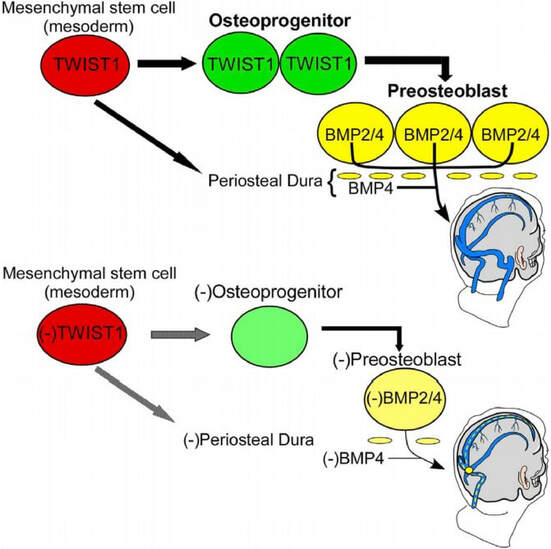
Background: Children born with skull malformations (craniosynostosis) often have cerebrovascular anomalies and increased intracranial pressure (ICP) that can lead to neurological complications if left untreated. Our lab characterized spectrum of dural cerebral vein malformations in TWIST1 mutation-positive individuals with craniosynostosis and signs of increased intracranial pressure (Tischfield et al., 2017). We also modeled cerebral vein development in mouse against classic anatomical descriptions of humans published in 1918, establishing mouse as a model for studying human cerebral vein malformations (see illustration on left). Surprisingly, conditional ablation of Twist1 via an endothelial-specific Cre-recombinase (Tie2) does not affect vascular development. Rather, Twist1 indirectly regulates the growth and remodeling of the dural cerebral veins by promoting osteoprogenitor cell development from coronal suture-derived mesodermal stem cells. These cells are capable of maturing into preosteoblasts that produce Bmp2 and Bmp4, and conditional inactivation of Bmp2/Bmp4 in coronal suture derived preosteoblasts and dura results in craniosynostosis and dural cerebral vein malformations. Furthermore, dural cerebral vein hypoplasia also results from loss of type II BMP receptor (BMPR2) signaling in endothelial cells, supporting the hypothesis that paracrine BMP signaling from developing skull tissue and/or dura is critical for venous angiogenesis in humans. Remarkably, venous malformations are not associated with arterial defects in TWIST1 mutation positive humans and mouse models, suggesting the head has evolved specialized signaling pathways that mediate tissue-specific angiogenic events. Collectively, this study represents the first characterization of cellular and molecular mechanisms that regulate the growth and remodeling of the large cerebral veins (Tischfield et al., 2017). It also suggests that pathways under the control of TWIST1 and/or BMP signaling may be necessary for proper blood and lymphatic vessel development in dura and other tissues, which is a current focus in the lab.
New and exciting directions in the lab: Investigate the development and functions of meningeal lymphatic vessels and the brain's glymphatic system: We are currently investigating cellular and molecular mechanisms that regulate the patterning of meningeal lymphatic networks, and how these processes may be affected in human craniofacial disorder models. We also wish to understand how these vascular networks integrate with and facilitate brain perivascular waste clearance pathways, known as the glymphatic system, and their involvement in human disease.
|
Cortical-striatal-thalamic development, function, and the pathogenesis of Tourette Syndrome
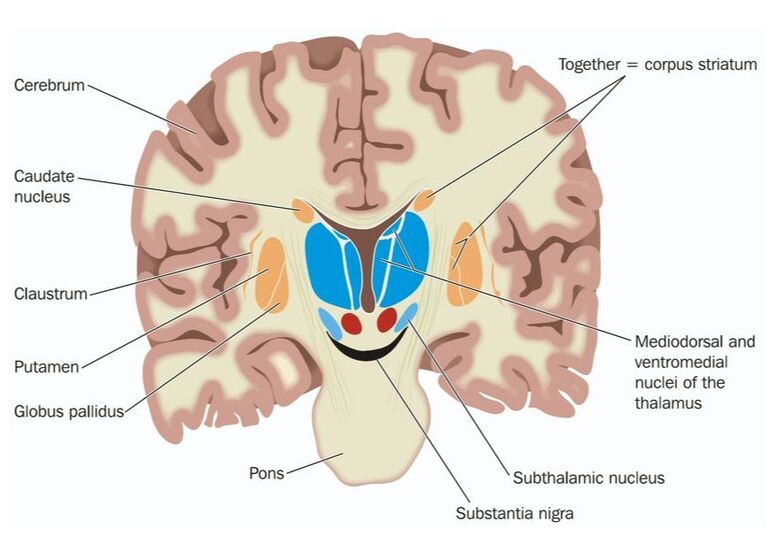
Background: Tourette Syndrome (TS) is a relatively common neurodevelopmental disorder that is estimated to occur in 0.5-1% of the general population. Similar to autism, TS is approximately four times as prevalent in males versus females. TS is characterized by the involuntary production of both motor and phonic tics, and diagnosis is made after observing both types of tics within a year. Both the types and severity of the motor and phonic tics can vary between different affected individuals. Simple motor tics typically involve eye blinking, head bobbing, and facial grimacing, but can also manifest as more complex motor movements (hand wriggling/twisting, jumping) that incorporate multiple muscle groups. Similarly, simple phonic tics may include persistent throat clearing or grunting, whereas complex vocal tics typically manifest as words or phrases. In extreme cases, vocal tics often include socially inappropriate words and phrases (coprolalia) that can pose significant social stigma and embarrassment during school and professional life. Tic behavior usually starts in young children and peaks during the teenage years, and most see improvement by early adulthood. For those at the extreme edges of the TS spectrum (10-15%), tics can remain chronic and debilitating throughout adulthood. In addition, approximately 40-50% of individuals with TS also have other neurological and behavioral problems such as obsessive-compulsive disorder (OCD) or attention-deficit/hyperactivity disorder (ADHD). These additional behavioral disturbances may persist even as motor and vocal tics wane. Treatment: Those who have mild forms of TS may not require medication if the tics do not interfere with routine daily life. However, people with debilitating and severe TS may require medication to help suppress tics, and neuroleptic drugs such as haloperidol may be administered. For those who have additional behavioral problems such as OCD or ADHD, methylphenidate or serotonin-specific reuptake inhibitors may be prescribed. Although current pharmacological approaches may lessen the frequency and severity of the tics, they do not usually completely eliminate the symptoms and can have significant side effects. For this reason, more research is necessary to identify and characterize the affected brain regions and neural circuitry of TS in order to devise more streamlined and effective pharmacological approaches. Neuropathology: The underlying neuropathology of TS remains poorly understood. Post-mortem brain tissue and functional magnetic resonance imaging (fMRI) imply combinatorial effects from many different brain regions, including the motor and sensorimotor cortices and their connections with the striatum. Interestingly, studies in rats and non-human primates suggest that functional subdivisions within the striatum and basal ganglia (illustrated above), and their connections with cortical, thalamic, and/or limbic areas can be parsed out to study the generation of motor versus vocal tics. Genetics: Few gene mutations, limited to only a small number of people in individual families, have been reported in TS. Recently, additional candidate TS genes were identified using whole-exome sequencing and de novo variant detection in 511 trios (Neuron. 2017 May 3;94(3):486-499), which consist of an unaffected mother and father plus the affected child (Tourette International Collaborative Genetics; Tourette Syndrome Association International Consortium on Genetics).
Our approach: In an effort to better understand the pathogenesis of TS, we have used CRISPR-Cas9 gene editing to engineer several TS mouse models that possess different point-mutations in high-confidence TS risk genes. With these genetically tractable models, our goal is to identify and characterize neuroanatomical and functional changes to brain development at both the single cell and circuit level to elucidate how brain dysfunction correlates with tics and behavioral abnormalities in TS. Towards this goal, we aim to implement a systems biology approach that utilizes operant conditioning, in vivo recordings and fiber photometry, electrophysiology/optogenetics, plus advanced behavioral paradigms that use machine learning algorithms.
Current Projects in the lab: We are using our novel mouse models to pursue several promising directions that will elucidate circuit mechanisms that underlie Tourette Syndrome. These include investigating changes to dopamine fluctuations within different striatal sub-compartments in freely behaving mice, and how this effects cognitive and sensorimotor functions. We are very interested in understanding the intersection between altered striatal dopamine, repetitive motor behaviors, and habit formation in mice. We are also interested in understanding perturbations to thalamocortical circuitry and how human mutations affect the formation of sensory maps in the somatosensory cortex.